Leave message
Can’t find what you’re looking for?
Fill out this form to inquire about our custom protein services!
Inquire about our Custom Services >>






























 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free!Get your ComboX free sample to test now!
Time Limited Offer: Welcome Gift for New Customers ! Shipping Price Reduction for EU Regions
> Insights > Amyotrophic Lateral Sclerosis: Pathology, Epidemiology, Pathogenesis and Therapeutic Drugs Aneuro
As a leading manufacturer of recombinant proteins and other critical reagents for support in developing target therapeutics, vaccines, and diagnostics, ACROBiosystems employs an application-oriented development strategy, with a particular focus on product design, quality control, and solution-based support.
Aneuro is a new product line of ACRO that focuses and reflects dedicated efforts in neuroscience research. We aim to promote and facilitate neuroscience research by providing high-quality protein products and valuable new ideas.

Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder with a median survival of 2 to 3 years from symptom onset.
ALS primarily affects the upper and lower motor neurons, but neurons in the frontal cortex and other neuroanatomical areas may also be affected. Loss of lower motor neurons from the spinal cord to the muscles leads to muscle weakness, atrophy, spasms, and fasciculation. Loss of upper motor neurons in the brain results in spasms, clumsiness, impaired reflexes, and functional limitations. Some patients also suffer varying degrees of extra-motor symptoms, such as cognitive and behavioral symptoms. The most common clinical manifestation is weakness and atrophy of the muscles in the hands, as if the body were gradually frozen.
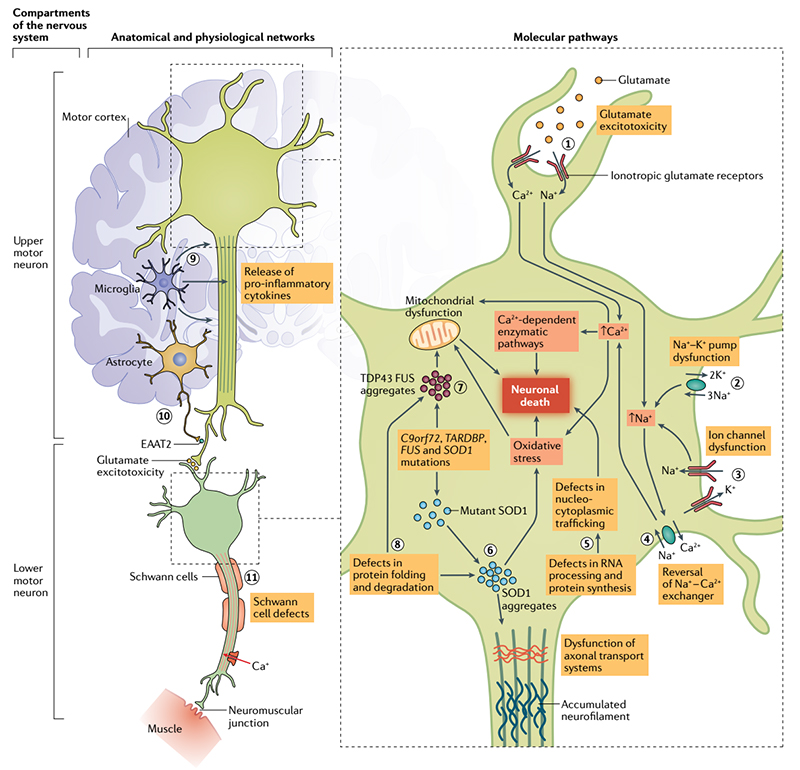
Pathophysiological mechanism of ALS
Clinical findings show that the incidence of the disease is significantly higher in people with a family history, and the combination of genetic and environmental factors increases the risk of sporadic ALS. The US National ALS Registry Act has substantially added to our overall knowledge about the epidemiology of ALS, and it estimates the ALS prevalence rate in the United States as 5 cases per 100,000 population for 2013. Approximately 10% of ALS is familial and caused by a genetic mutation that is usually inherited in a mendelian autosomal dominant manner. A hexanucleotide G4C2 repeat expansion in the chromosome 9 open reading frame 72 gene (C9orf72) is the most commonly known genetic cause of ALS, accounting for 30% to 40% of familial ALS, and it also causes frontotemporal dementia (FTD). Together with the C9orf72 repeat expansion, mutations in genes encoding copper-zinc superoxide dismutase (SOD1), transactive response DNA-binding protein 43 (TDP-43), and fused in sarcoma (FUS) account for more than 50% of the familial ALS cases, and another 30 or so genes have been identified as possibly causing ALS.
At present, the pathogenesis of ALS is still unclear, but the pathological features and gene mutations related to ALS have provided important clues to the etiology of ALS. A common feature of many neurodegenerative diseases is the accumulation of proteins in the cells that make up the nervous system leading to cell dysfunction and even death.
Clinicopathologically, 97% of ALS patients have brain ectopic aggregation of TDP-43 (transfer from nucleus to cytoplasm and formation of protein aggregation), and cellular and animal studies have confirmed the neuronal toxicity of this pathological TDP-43. Therefore, studying the mechanism related to pathological TDP-43 has become an important direction to conquer ALS. A feed-forward loop model involving oxidative stress is proposed in a study published in Nature Structural & Molecular Biology in 2021. In this study, it was suggested that TDP-43 nucleates accumulate in the cytoplasm. On the one hand, by capturing microRNA, the inhibitory effect of downstream target mRNAs was weakened and the expression of some mitochondrial genes was enhanced. On the other hand, specific mitochondrial proteins were induced to co-agglomerate, resulting in their loss of function. This results in an imbalance of mitochondrial structure and function, resulting in increased ROS and further enhanced TDP-43 truncation and aggregation. This positive feed cycle reveals that the interaction between oxidative stress and specific gene mutations may be one of the important mechanisms for the initiation and development of ALS.
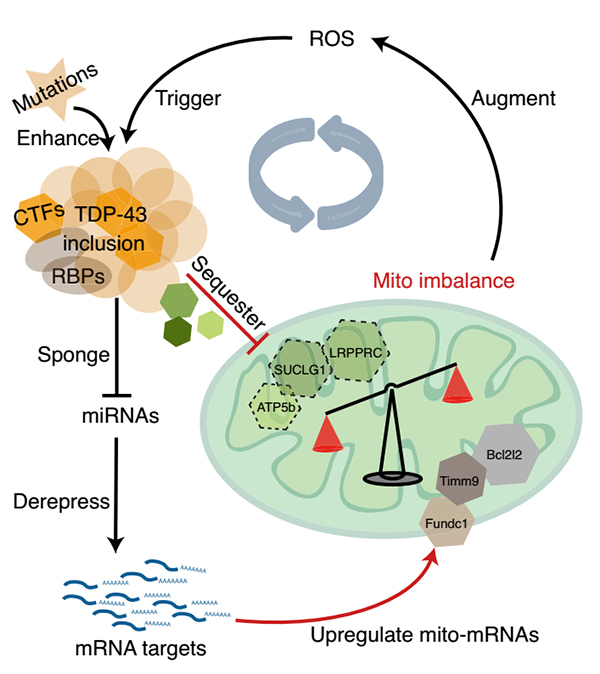
model of feed-forward loop mitochondrial imbalance caused by TDP-43 aggregation
Triggering receptor expressed on myeloid cells 2 (TREM2), which is only expressed in microglia of the central nervous system. Mutations of TREM2 are involved in Alzheimer's disease, Parkinson's disease (PD) and a variety of other neurodegenerative diseases. There are also clinical reports that TREM2 mutations also increase the risk of ALS. A 2021 study published in Nature Neuroscience is the first to demonstrate a protective role of microglia TREM2 in TDP-43 related neurodegenerative diseases (not limited to ALS) and the first to report TDP-43 as a ligand of microglia TREM2. Based on this, a modest increase in microglia TREM2 expression and activity at specific disease stages may help alleviate ALS-related dysfunction. A single dose of anti-TREM2 mAb has been tested in an animal model of AD, promoting microglial metabolic activation and proliferation, thereby enhancing the phagocytosis of amyloid beta (Aβ).
Riluzole, a glutamate receptor antagonist, was approved by the FDA in 1995 for the treatment of ALS. In 2017, the FDA approved a free radical scavenger originally used for acute ischemic stroke and a powerful antioxidant, edaravone, for the treatment of ALS. In addition to Ezogabine, Rasagiline, researchers are also actively exploring small molecule drugs, genetic pathogenesis, biological drugs and cell therapy to develop drugs for the treatment of ALS.
Aneuro focuses on neuroscience and provides SOD1 and TREM2 proteins for therapeutic drug development for ALS.
| Molecule | Cat. No. | Species | Product Description |
|---|---|---|---|
| SOD1 | SO1-H5148 | Human | Human SOD1 / Cu-Zn SOD Protein, His Tag |
| TREM2 | TR2-H5254 | Human | Human TREM2 Protein, Fc Tag |
| TR2-H5256 | Human | Human TREM2 Protein, Mouse IgG2a Fc Tag | |
| TR2-H82E7 | Human | Biotinylated Human TREM2 Protein, His,Avitag™ | |
| TR2-H52H5 | Human | Human TREM2 Protein, His Tag | |
| TR2-M52H3 | Mouse | Mouse TREM2 Protein, His Tag | |
| TR2-M5254 | Mouse | Mouse TREM2 Protein, Fc Tag | |
| TR2-C52H3 | Cynomolgus | Cynomolgus TREM2 Protein, His Tag |
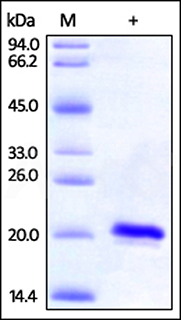
Human SOD1, His Tag (Cat. No. SO1-H5148) on SDS-PAGE under reducing (R) condition. The gel was stained overnight with Coomassie Blue. The purity of the protein is greater than 97%.
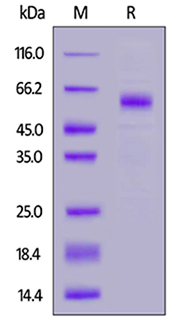
Human TREM2, Fc Tag (Cat. No. TR2-H5254) on SDS-PAGE under reducing (R) condition. The gel was stained overnight with Coomassie Blue. The purity of the protein is greater than 95%.
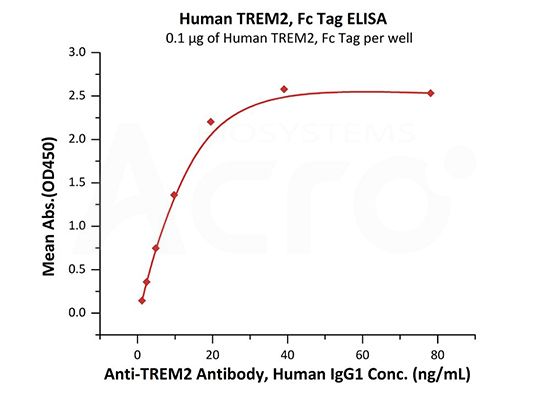
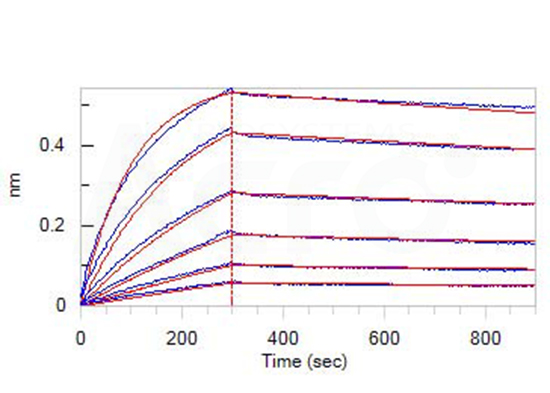
Immobilized Human TREM2, Fc Tag (Cat. No. TR2-H5254) at 1 μg/mL (100 μL/well) can bind Anti-TREM2 Antibody, Human IgG1 with a linear range of 1-20 ng/mL (QC tested).
>>>Learn more about Aneuro: advancing neuroscience research
ACRO is developing more proteins for AD diagnosis and treatment. If you have any needs or more development suggestions, don't hesitate to contact us.
1. Kiernan MC, Vucic S, Talbot K, McDermott CJ, Hardiman O, Shefner JM, Al-Chalabi A, Huynh W, Cudkowicz M, Talman P, Van den Berg LH, Dharmadasa T, Wicks P, Reilly C, Turner MR. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol. 2021 Feb;17(2):104-118.
2. Oskarsson B, Gendron TF, Staff NP. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin Proc. 2018 Nov;93(11):1617-1628.
3. Zuo X, Zhou J, Li Y, Wu K, Chen Z, Luo Z, Zhang X, Liang Y, Esteban MA, Zhou Y, Fu XD. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat Struct Mol Biol. 2021 Feb;28(2):132-142.
4. Xie M, Zhao S, Bosco DB, Nguyen A, Wu LJ. Microglial TREM2 in amyotrophic lateral sclerosis. Dev Neurobiol. 2022 Jan;82(1):125-137.
5. Xie M, Liu YU, Zhao S, Zhang L, Bosco DB, Pang YP, Zhong J, Sheth U, Martens YA, Zhao N, Liu CC, Zhuang Y, Wang L, Dickson DW, Mattson MP, Bu G, Wu LJ. TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration. Nat Neurosci. 2022 Jan;25(1):26-38
This web search service is supported by Google Inc.
















 A-Z
A-Z