


Leave message
Can’t find what you’re looking for?
Fill out this form to inquire about our custom protein services!
Inquire about our Custom Services >>






























 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free!Get your ComboX free sample to test now!
Time Limited Offer: Welcome Gift for New Customers ! Shipping Price Reduction for EU Regions
> Insights > Special Topic on Deep Interpretation of GMP Product Quality--Topic 3

In recent years, the biopharmaceutical industry has witnessed a remarkable growth, with cell and gene therapy emerging as a pivotal sector amidst the continuous technological advancements. As biologics capture a larger share of next-generation medicine, ensuring these biological medicines are safe has become the utmost priority. This brings further scrutiny not only to the final cell and gene therapy, but also to the raw materials used during manufacturing and production. This has been further underscored by the FDA emphasizing the use of ‘the highest quality’ materials suitable. Similarly, the International Pharmaceutical Regulatory Plan Cell Therapy Working Group drafted a deliberative document disseminating their perspective on using high quality raw materials in manufacturing and licensed human CGT products.
With regulatory bodies emphasizing the use of ‘high quality’ raw materials, what does this mean for cell therapy manufacturers?
In short, cell therapy manufacturers must do their due diligence in ensuring the quality of their raw materials, whether it comes from a supplier or developed in-house.
Evaluations on quality control methods and standards for relevant raw materials including cytokines and other growth factors, are a necessary step within an investigational new drug (IND) application before proceeding with clinical research. These methods and standards are derived from several sources. Pharmacopeial methods, derived from organizations such as USP or the European Directorate for the Quality of Medicines & Healthcare (EDQM) are standardized approaches that establish a fundamental requirement for production and release. Non-pharmacopeial methods, such as the International Conference on Harmonization (ICH) documents, are supplemental analytical approaches tailored towards evaluating specific characteristics of a product and its manufacturing process.
Many commercial products are currently marketed to follow these standards, commonly labeled ‘GMP-grade’ or ‘cGMP-grade’. However, products that comply with the stated standards are often self-regulated. As such, responsibility still falls upon cell therapy manufacturers to perform due diligence, which can make it difficult to find a trustworthy supplier.
Herein, we highlight the three main aspects of a GMP-grade raw material quality control system that are critical towards meeting IND requirements and successfully moving into the clinical phase.
Evaluating raw material safety is defined through various contaminant controls and detection strategies to limit any residues that could cause harm or affect the final therapeutic. This includes sterility tests, evaluations of contaminants such as endotoxins, mycoplasma, residual host cell DNA, and residual host cell proteins.
• USP<71>Sterility Tests
Ensuring the sterility of final cell and gene therapies is tricky. Since the final therapeutic is cells, there is no easy method such as sterile filtration, autoclaving, etc. Thus, manufacturers must be very careful throughout the manufacturing process to ensure sterility of the final product. This applies to the raw materials used as well, requiring manufacturers to evaluate suppliers stringently. As such, raw materials suppliers for GMP-grade manufacturers including ourselves, are expected to control cytokines and other growth factors stringently, incorporating automatic fill-finish in B+A cleanrooms, online environmental monitors, and USP-compliant sterility tests. Thus, evaluating for sterility of raw materials is the primary methodology to ensuring sterility of the final therapeutic. For more information, read our Special Topic on Deep Interpretation of GMP Product Quality – Topic 2.
• USP<85>Endotoxin Tests
Endotoxins are toxins present in bacterial cells that are released after cell disintegration which can sometimes lead to diseases such as botulism. Maintaining a low endotoxin level in biologics is essential for ensuring patient safety and preventing endotoxin-related diseases. Several methods are acceptable: qualitative gel clot and chromogenic LAL-based methods. However, if there are any inconsistent or conflicting results, the gel clot result is deemed authoritative. Internally, we utilize a LAL method calibrated against USP Reference Standard endotoxin calibrants to ensure accuracy with sample results shown in Table 1. Subsequently, to ensure endotoxin testing method is valid, preparatory testing as outlined in USP<85>was performed, with an acceptable endotoxin recovery rate defined as being within the range of 50 to 200%.
Table 1. Endotoxin Sample Curve Evaluation
| Theoretical value | Detected value | Recovery rate |
|---|---|---|
| 50 EU/ml | 55.04 EU/ml | 113.0% |
| 5 EU/ml | 5.96 EU/ml | 104.7% |
| 0.5 EU/ml | 0.6 EU/ml | 108.5% |
| 0.05 EU/ml | 0.06 EU/ml | 108.0% |
• USP<63>Mycoplasma Tests
Mycoplasma is a common contaminant in cell and tissue cultures that results in the alteration of cellular growth and phenotype. Testing for mycoplasma is critical in ensuring reliability in biologics and its raw materials. When evaluating mycoplasma, the gold standard remains as the culturing method, monitoring for growth of typical mycoplasma colonies on solid media. Other validated methodologies include the use of fluorescent dyes to highlight characteristic particulates or filamentous patterns on the cell surface. Although nucleic acid amplification techniques or enzyme activity-based methods can also be used, suitable validation and comparisons between methodologies to cell culture must also be addressed.
• USP<509, 1132>Residual Host Cell DNA and Proteins
Host cell DNA and protein are other factors that must also be controlled to acceptable levels to avoid any safety risks. Foreign DNA and protein are susceptible to causing immunogenic and oncogenic reactions that could cause more harm. Ensuring that these impurities are removed in any raw materials and subsequent manufacturing processes is crucial to patient safety.
Of course, as with any testing or analytical method, validation is necessary to ensure that the tests used are accurate. The USP documents referenced above provide assay protocols and clear criteria for evaluating tests that are standardized across biologic materials. However, when it comes to material-specific assessments such as bioactivity, analytical methods must first be validated, most commonly following ICH Q2 (Revision 1), titled “Validation of Analytical Procedures Text and Methodology”.
Table 2. Validation Characteristics for Consideration
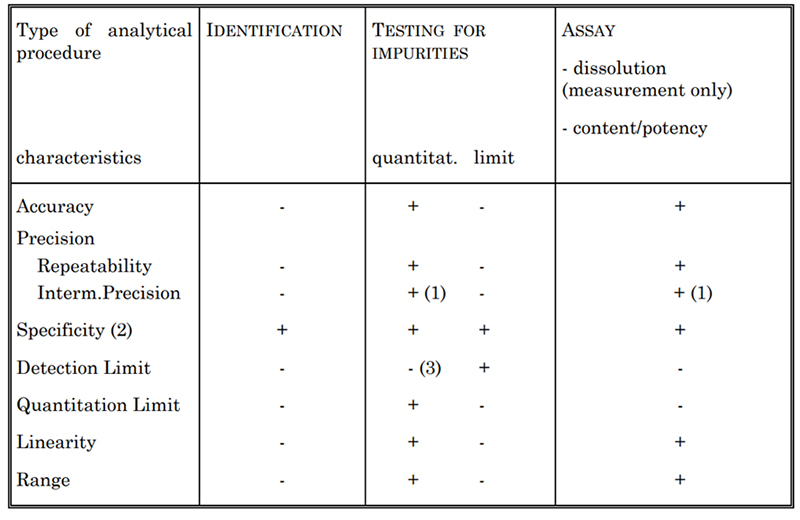
Depending on the type of assay to be validated, certain characteristics are generally assessed, as listed in Table 2. This includes specificity, accuracy, precision, detection limit, quantitation limit, linearity, and range. For example, to determine cytokine concentration, we establish different assay methods, such as US-Spectrophotometric assay and the Lowry method. Each of these methods are fully validated in accordance with USP and ICH Q2 guidelines.
Table 3. Validation of UV-Spectrophotometric Assay
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Accuracy | Recovery Rate: 96% | Recovery Rate: 90%-108% | Pass |
| Repeatability | RSD: 0.05% | RSD≤3% | Passtd> |
| Intermediate Precision | RSD: 0.48% | RSD≤3% | Pass |
| Robustness | RSD: 0.04% | RSD≤3% | Pass |
| Linearity | R2: 0.99925 | R2>0.999 | Pass |
| Range | 0.0452-0.452 mg/ml | 0.0452-0.452 mg/ml | Pass |
To further validate the UV-spectrophotometric assay, a secondary confirmatory method, the Lowry method, was used. As a mature, well-established methodology, the Lowry method provides a solid foundation for comparison. Thus, direct comparisons of the quantitative result of six different spiked samples ensure the accuracy and reliability of a UV method, shown in Table 4.
Table 4. Secondary Confirmation Assay – Lowry Method
| Method Validation | Lowry | UV Method | Conclusion |
|---|---|---|---|
| Quantitative Result | 461 ug/ml | 436 ug/ml | Pass |
| 476 ug/ml | 436 ug/ml | ||
| 465 ug/ml | 436 ug/ml | ||
| 472 ug/ml | 436 ug/ml | ||
| 468 ug/ml | 436 ug/ml | ||
| 461 ug/ml | 435 ug/ml |
Using the same analytical method validation framework, this includes other assays such as cell activity. For example, our TNF-α assay includes accuracy, intermediate precision, linearity, and range. However, the criteria differ due to the purpose of the assay.
Table 5 Example of TNF-α Cell activity validation
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Relative Accuracy |
Bias:1.1% Slope:1.01 |
Bias within ±12% range & Slope of regression equation between 0.80 and 1.25 | Pass |
| Intermediate Precision | GCV*:11.2% | (GCV)* ≤ 20% | |
| Specificity | Difference % (buffer): 8.8% | Difference % (buffer) ≤ ±10% | |
| Linearity | Correlation Coefficient: 0.97 | Correlation Coefficient ≥ 0.95 | |
| Potency Range | Potency Range: 64% -156% | Range of product efficacy standards (64% -156%) |
*GCV is calculated as the anti-log of the standard deviation. GCV = anti-log(SD)
Stability testing is the basis of evaluating a product’s shelf-life. As such, this testing is a critical component throughout the entire drug development, clinical, market launch, and post-market monitorization in quality research processes purposes. This means stability testing must be conducted based on the unique qualities and characteristics of the product. Additionally, performing stability studies systematically ensures minimal lot-to-lot variation, while also providing clear instructions for storage upon delivery.
To evaluate stability and consistency within a viable timeframe, the accelerated stability testing method, based on the Arrhenius equation, is a widely recognized strategy, and numerous studies in recent years have demonstrated its applicability and accuracy. Both the 2002 document "EN13640 In Vitro Diagnostic Medical Devices Stability Testing" published by the European Committee for Standardization and the 2009 document "EP25A Evaluation of Stability of In Vitro Diagnostic Reagents" released by the Clinical and Laboratory Standards Institute (CLSI) recommend the use of this method for determining the stability of in vitro diagnostic reagents. Varying stability assessment parameters are shown in Table 6.
Table 6. Stability Assessment Parameters

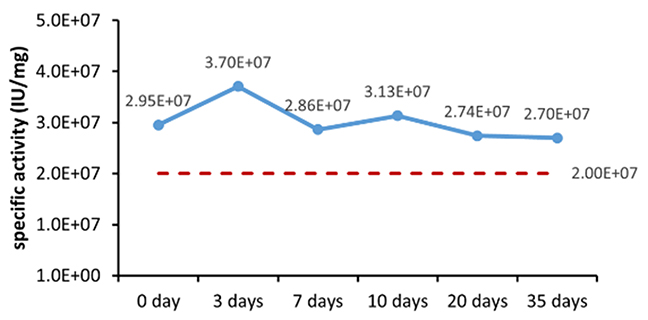
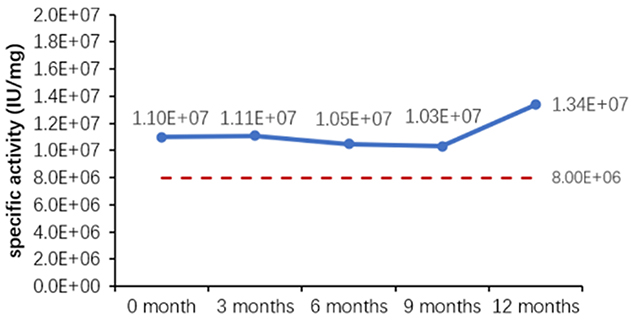
Figure 1. (A) Accelerated and (B) real-time stability evaluations of finished product (powder).
Quality control evaluations generally opt for more mature and sensitive that meets specified requirements. Due to the potential limitations in analytical methods, it is advisable to consider utilizing simultaneous complementary methods to comprehensively evaluate not only raw materials, but also the final therapeutic. The analytical methods themselves also need to undergo rigorous validation. A well-established, stringent, and professional quality control system can be what makes-or-breaks any therapeutic approval process and should reference pharmacopeia standards such as USP. Thus, the international perspective on pharmaceutical quality management has evolved from drug quality controlled through inspection" to "drug quality is achieved through process control in production" and finally to, "drug quality is produced through good design (Quality by Design, QbD)." Embracing this QbD philosophy implies establishing a correlation between product quality attributes and clinical safety and efficacy, necessitating the creation of a comprehensive quality control system throughout the entire process and product lifecycle management. As such, it is through this mindset that ACROBiosystems continuously refines our GMP quality management system to help provide reliable support when it comes to raw materials for cell and gene therapies.





This web search service is supported by Google Inc.
















 A-Z
A-Z












